利用傅里叶变换红外光谱法测定甲酸的二聚热
Giles Henderson
东伊利诺伊大学,查尔斯顿,伊利诺伊州 61920
尽管微型计算机在大学中日益普及,但典型的大学物理化学实验教材并未描述能充分利用其能力的实验。本文将描述如何根据二聚体平衡常数的温度依赖性来测定甲酸的二聚热。实验中采用计算机对红外干涉图进行傅里叶变换得到透射光谱,将透射率转换为吸光度,进行光谱扩展,通过光谱乘法和光谱减法分离单体和二聚体的贡献,并通过光谱积分确定蒸气组分,此外还进行最小二乘拟合和数字绘图。尽管该项目是使用配备商业软件的 Nicolet 20DXB 型傅里叶变换红外光谱仪完成的,但只要与微型计算机连接,该测量也可以轻松地用传统色散型仪器进行(1)。
甲酸二聚体形成的热力学
羧酸分子即使在气相中也存在强烈的缔合作用。早期的电子衍射研究(2)表明,甲酸蒸气中富含一种由两个氢键稳定的平面二聚体形式(见图1)。单体-二聚体平衡,
2CH2O2⇄(CH2O2)2+ 热 (1)
可用平衡常数表示
KD=P(CH2O2)2/P2(CH2O2)(2)
其中组分 i 的活度由其分压 Pi 给出。预计蒸气的红外光谱将表现为单体和二聚体特征峰重叠的混合谱。
从方程 2 可以明显看出,二聚体的浓度将随单体分压的平方而增加。因此,样品压力的变化预计会引起单体与二聚体比例的变化。这提示了通过适当的数据处理来分离重叠的单体和二聚体****光谱的可能性。
由于二聚体的形成是放热过程,因此平衡(方程1)预计会向左移动,即在高温下有利于高浓度的单体和低浓度的二聚体。如果单体和二聚体的特征峰能够完全分辨,并且已知相应的比尔定律****积分吸收系数,那么平衡混合物的组成和平衡常数的值就可以通过红外吸收光谱实验确定。假设蒸气组分为理想气体,则 Pi=[i]RT。
 图 1. 甲酸二聚体的 C2h 结构。
图 1. 甲酸二聚体的 C2h 结构。
 图 2. 甲酸蒸气在 20∘C 和约 0.01 atm 下的傅里叶变换红外吸收光谱。该光谱使用 10 cm KBr 吸收池,扫描 20 次,分辨率为 4−cm−1。
图 2. 甲酸蒸气在 20∘C 和约 0.01 atm 下的傅里叶变换红外吸收光谱。该光谱使用 10 cm KBr 吸收池,扫描 20 次,分辨率为 4−cm−1。
KD=[CH2O2]2RT[(CH2O2)2]=⟨M⟩2T⟨D⟩(⟨ϵD⟩R⟨ϵM⟩2l)(3)
其中 [i] 是组分 i 的摩尔浓度,⟨D⟩ 是二聚体一个完全分辨的红外谱带的积分吸收。⟨M⟩ 是一个分辨的单体谱带的积分吸收,⟨ϵD⟩ 和 ⟨ϵM⟩ 分别是二聚体和单体相应的积分吸收系数,l 是光程,而 R 是气体常数。此外,KD 将根据 ΔHD 的大小随温度变化:
ΔGD∘=−RTln(KD)=ΔHD∘−TΔSD∘(4)
其中 ΔGD∘,ΔHD∘ 和 ΔSD∘ 分别是二聚反应的标准自由能、焓和熵。如果我们将方程 3 代入 KD 并重新整理,可以很容易地得到
ln(⟨M⟩2T⟨D⟩)=R−ΔHD∘(T1)+[RΔSD∘−ln(⟨ϵD⟩R⟨ϵM⟩2l)](5)
因此,只需要知道吸收光谱的温度依赖性,就可以确定 ΔHD∘ 以及二聚体中的氢键能。
实验部分
将几滴试剂级甲酸(Fisher A-118)放入一个配有水银压力计和液氮冷阱的真空管路贮存器中。使用一根两端配有球形接头的 1 米长派热克斯玻璃管将真空管路连接到一个 10 cm 长的 KBr 气体池。气体池用加热带包裹,并配备一个热敏电阻探头,以将池温维持在 0.1∘C 的精度。将气体池安装在 Nicolet 20DXB 型傅里叶变换红外光谱仪中。由于需要吹扫单光束干涉仪以排除 CO2 和 H2O 蒸气,因此在整个实验期间,样品池保持不动,其派热克斯连接管和电线被密封在已吹扫的样品仓内。将大约 0.01 atm 的甲酸蒸气转移到气体池中,并加入 N2 使总压达到 1 atm。图 2 展示了由 20 个干涉图经傅里叶变换得到的典型
 图 3. 甲酸蒸气光谱的选定部分。上方的谱线是在 0.010 atm 下扫描 20 次得到的,下方的谱线是在 0.0025 atm 下扫描 320 次得到的。图中给出了两个特征峰的积分吸收比,这使得可以对单体和二聚体的谱带进行归属。
图 3. 甲酸蒸气光谱的选定部分。上方的谱线是在 0.010 atm 下扫描 20 次得到的,下方的谱线是在 0.0025 atm 下扫描 320 次得到的。图中给出了两个特征峰的积分吸收比,这使得可以对单体和二聚体的谱带进行归属。
 图 4. 此处通过高压和低压蒸气光谱的适当线性组合,分别以“正残差光谱”和“负残差光谱”的形式获得了二聚体和单体的吸收光谱。
图 4. 此处通过高压和低压蒸气光谱的适当线性组合,分别以“正残差光谱”和“负残差光谱”的形式获得了二聚体和单体的吸收光谱。
4−cm−1 分辨率光谱。然后将样品稀释至约原浓度的 1/4,并采集 320 次扫描的光谱。扫描次数增加了 (4)2 倍,以保持与较浓样品相同的信噪比。在室温至 45.0∘C 范围内的五个不同温度下重复此过程,并将数字化光谱存储在软盘上以备后续数据处理。
数据处理
图 3 比较了两种不同压力下甲酸蒸气光谱选定部分的放大图。我们注意到,由于甲酸压力的增加,位于 1105 cm−1 的特征峰的积分吸收增加了 1.66 倍,而位于 1218 cm−1 的特征峰的积分吸收增加了 2.80≃(1.66)2 倍。1218−cm−1 谱带的二次方压力依赖性使我们能将其归属于二聚体,而将 1105−cm−1 的信号归属于单体。现在,如果我们将整个低压光谱乘以 1.66,然后从高压光谱中减去它,我们预计所有单体的特征峰都将被完全抵消,得到的正残差吸收将归因于二聚体。同样地,如果我们将整个低压光谱乘以 2.80,然后从高压光谱中减去它,所有二聚体的贡献都将被完全抵消。
甲酸吸收****蒸气组分
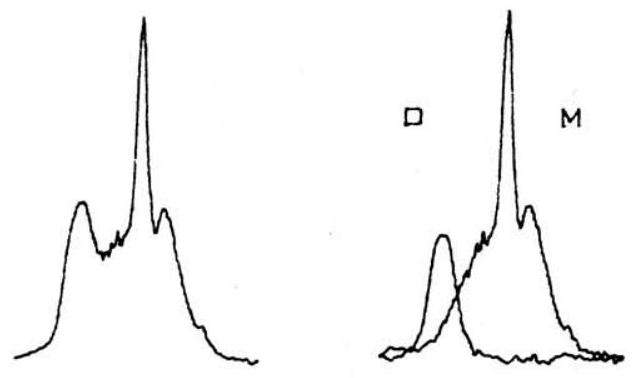

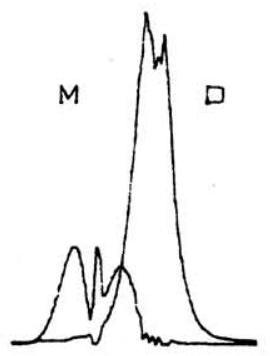
图 5. 这里描述的技术为分离重叠的吸收带提供了一种强大的数值方法。上方谱线给出了 760 至 530 cm−1 之间吸收的单体和二聚体组分,下方谱线给出了 1830 至 1680 cm−1 之间 C=O 伸缩振动模式的组分。
 图 6. 红外吸收的温度依赖性。单体的 C-O 伸缩振动带(位于 1105 cm−1)随温度升高而增强,而二聚体的 C-O 伸缩振动带(位于 1218 cm−1)则相应减弱。
图 6. 红外吸收的温度依赖性。单体的 C-O 伸缩振动带(位于 1105 cm−1)随温度升高而增强,而二聚体的 C-O 伸缩振动带(位于 1218 cm−1)则相应减弱。
然而,在这种情况下,我们将过度减去单体的贡献,因此会得到一个单体的“负残差光谱”。这两种结果都显示在图 4 中。我们注意到,如图 5 所示,单体和二聚体的特征峰得到了非常令人满意的分离,尤其是在谱带重叠的区域。当我们注意到图 2 中单体的 2945−cm−1 C-H 伸缩振动模式如何被重叠的二聚体吸收完全掩盖,而在图 4 中这个弱谱带又如何清晰地显现出来时,我们就能进一步体会到该技术的价值。
表 1. 甲酸吸收的温度依赖性 a
| t(∘C) |
Pb |
⟨ D ⟩1218 |
⟨M⟩1105 |
ln(⟨D⟩/⟨M⟩2T) |
| 29.1 |
h |
22.42 |
16.51 |
-8.21 |
| 29.1 |
l |
8.012 |
9.954 |
-8.23 |
| 33.0 |
h |
30.54 |
21.72 |
-8.46 |
| 33.0 |
l |
9.904 |
12.38 |
-8.46 |
| 37.0 |
h |
30.60 |
24.45 |
-8.71 |
| 37.0 |
l |
9.378 |
13.56 |
-8.71 |
| 41.0 |
h |
27.38 |
25.77 |
-8.94 |
| 41.0 |
l |
8.301 |
14.32 |
-8.96 |
| 45.0 |
h |
33.35 |
31.55 |
-9.16 |
| 45.0 |
l |
10.27 |
17.61 |
-9.17 |
a⟨D⟩ 和 ⟨M⟩ 分别对应于 1218 cm−1 处的 ν6 二聚体谱带(4)和 1105 cm−1 处的 ν6 单体谱带(3)的积分吸收。
b h 和 l 分别代表约 0.010 和 0.0025 atm 的甲酸蒸气。
 图 7. ΔH0 从方程 5 对观测数据的最小二乘拟合的斜率中获得。此处 Q=⟨D⟩/⟨M⟩2T。
图 7. ΔH0 从方程 5 对观测数据的最小二乘拟合的斜率中获得。此处 Q=⟨D⟩/⟨M⟩2T。
有好奇心的学生可能希望将这些观测到的振动频率与最近对单体(3)和二聚体(4)进行简正坐标分析所计算出的值进行比较。
既然我们有了一种能精确分离单体和二聚体****吸收的简单方法,我们就可以研究温度对平衡组成的影响。图 6 显示了在五个温度下二聚体(1218 cm−1)和单体(1105 cm−1)的 C-O 伸缩振动带。我们注意到,单体的吸收以二聚体吸收的减少为代价而增加,这与放热反应的预期相符。这些特征峰的积分吸收汇总于表 1。然后将这些结果用于方程 5 来获得二聚热。ln[⟨D⟩/(⟨M⟩2T)] 对 T−1 的作图见图 7。最小二乘斜率 (5.76±0.5)×103 乘以 −R 得到 ΔHD∘=−47.9±0.4 kJ/mol。该结果与表 2 中的其他文献值进行了比较。
讨论
从表 2 可以明显看出,关于甲酸的 ΔHD 值存在很大分歧。Coolidge (5) 进行的非常仔细的蒸气密度测量给出的实验结果高于吸收法测量的结果。这两种方法之间的差异或许是真实存在的,其原因在于蒸气密度法包含了所有类型的缔合,而吸收法则不然。Herman (6) 报告的 ΔHD 值略高于本文测定的结果。
表 2. 甲酸的二聚热
| 方法 |
年份 |
来源 |
ΔHb( kJ/mol) |
| 蒸气密度 |
1928 |
Coolidge (5) |
-59 |
| 红外吸收 |
1940 |
Herman (6) |
−52±3 |
| LCAO-MO-SCF |
1971 |
Clementi et al. (8) |
-67.8 |
| 最小基组-STO-SCF优化结构 |
1976 |
Del Bene et al. (9) |
-63.2 |
| 双泽塔SCF(含畸变能) |
1978 |
Smit et al. (10) |
-49.4 |
| FTIR |
本工作 |
|
−48.9±0.4 |
然而,这种差异可能是由于 Herman 使用了最大吸收波长 λmax 处的吸收值,而非积分吸收值。当温度变化时,相应转动能级的玻尔兹曼布居会发生变化,导致谱带形状不同。这种现象可能导致在 λmax 处的表观吸收系数随温度变化而变化,从而在 lnK 对 1/T 的斜率中引入系统误差。通过对整个观测到的转动包络进行吸收积分,可以很明显地避免这个问题。Clague 和 Bernstein (7) 比较了这两种方法,同样观察到当使用谱带面积而非峰高时,ΔHD 会降低 4−kJ/mol。
我们还注意到理论从头计算值(8-10)之间也存在巨大差异。然而,随着基组和优化几何构型的不断改进,这些计算结果正逐渐与实验光谱值趋于一致。
从方程 5 可以看出,如果已知积分吸收系数的精确值,就可以从图 7 的截距中确定 ΔSDo,精确到三位有效数字。这将需要精确的样品浓度,或者进行比简单水银压力计所能达到的精度高一个数量级的压力测量。另一种方法是在足够高的温度下测量光谱,此时样品几乎 100% 解离,从而可以直接根据比尔定律确定 ⟨ϵM⟩。在此,我们根据最小二乘截距(−27.3±0.2)和吸收系数的合理限值,估计 ΔSD∘ 为 −180±50 J/moldeg。ΔS 的符号证实了在二聚体形成过程中熵或无序度的减少。
总而言之,这项傅里叶变换红外光谱测量似乎非常适合现代大学物理化学实验室。它利用微型计算机的能力来完成那些否则会因计算量过大而无法进行的计算,并同时阐释了基本的热力学平衡原理。该练习可以轻松地在两个 3 小时的实验课时内完成。或许,最高效的设备使用方式是在第一个课时收集一组数据,将所有光谱存储在软盘上,然后在第二个课时安排个别学生(或两人一组)进行键盘操作以完成数据处理。
致谢
作者希望对东伊利诺伊大学教师研究委员会为本项目提供的资金支持表示感谢。
参考文献
(1) Mattson, B. M.; Shepherd, T. R.; Solsky, J. F. J. Chem. Educ. 1985, 62, 690.
(2) Karl, J.; Brockway, L. J. Amer. Chem. Soc. 1944, 66, 574.
(3) Redington, R. J. Mol. Spectrosc. 1977, 65, 171.
(4) Hagen, I.; Cyvin, S. J. Mol. Struct. 1971, 8, 159.
(5) Coolidge, A. J. Amer. Chem. Soc. 1928, 50, 2166.
(6) Herman, R. J. Chem. Phys. 1940, 8, 252.
(7) Clague, A.; Bernstein, H. Spectrochim. Acta 1969, 25A, 593.
(8) Clementi, E.; Mehl, J.; von Niessen, W. J. Chem. Phys. 1971, 54, 508.
(9) Del Bene, J.; Kochenour, W. J. Amer. Chem. Soc. 1976, 98, 2041.
(10) Smit, P.; Derissen, J.; van Duijneveldt, F. J. Chem. Phys. 1978, 69, 4241.